Anemia due to hemolysis Flashcards
Define hemolysis and describe the two main mechanisms of increased destruction of RBCs: intravascular and extravascular hemolysis.

Hemolysis is defined as a decrease in red cell survival or increase in turnover beyond standard values. The pace or rate of hemolysis, in part, determines whether anemia presents acutely or over a more insidious, chronic course. The degree of anemia is affected by the extent to which marrow production is increased. Increased production may compensate for the increased turnover, which results in no or mild anemia. Or may not be able to keep up with red cell destruction (uncompensated, moderate to severe anemia).
Intravascular: turnover within the vascular space (in the blood). Red cells undergoing intravascular hemolysis release hemoglobin into the circulation. The tetramer form of hemoglobin is unstable and dissociates into αβ dimers which may be immediately bind to haptoglobin. This complex is removed from the circulation by the liver. Although haptoglobin has a very high affinity and specificity for hemoglobin, its capacity may be easily overwhelmed (this leads to a decrease in haptoglobin) by significant intravascular hemolysis leaving the released hemoglobin to be broken down by other pathways.
The iron in hemoglobin can be oxidized to form methemoglobin. Dissociation of globin releases metheme which may bind to albumin or hemopexin. These latter compounds may be taken up by hepatic parenchymal cells and converted to–> bilirubin. Alternately, the monomeric forms of methemoglobin or hemoglobin may be filtered and not reabsorbed by the kidney and appear in the urine.
A decrease in serum haptoglobin levels, detection of hemoglobin in the urine or plasma and increase in metheme or methemalbumin all suggest intravascular hemolysis.

Explain extravascular hemolysis:
With extravascular hemolysis, the red cell is ingested by macrophages of the RE system.
- The heme is separated from globin, iron removed and stored in ferritin (can be tranferred into the plasma with transferrin), and the porphyrin ring converted to bilirubin which is released from the cell.
- Taken up by a specific transport system in the liver, the bilirubin (lipid soluble) is converted to a water soluble compound by addition (conjugation) of a glucuronic acid. This is completed by the cytochrome P-450 enzyme(s) in liver parenchymal cells. After secretion into the biliary tract and small bowel, the glucuronic acid is removed and bilirubin converted into urobilinogen and other water soluble pigments. Urobilinogen may cycle between the gut and liver (entero-hepatic circulation) or excreted by the kidney into the urine.
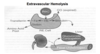
RBC mechanics:
What factors promote survival of RBCs?
- Normal RBC lifespan = 120 days
- Hemolysis = Decreased RBC lifespan due to premature RBC destruction/increased RBC turnover
- Reticulocyte production can increase by 6-8 fold in response to increased erythropoietin secretion.
•If RBC destruction >> RBC production -> hemolytic anemia
- *Characteristics of RBCs promoting survival:**
- ->Deformable membrane and cytoskeleton
- ->Optimal surface-to-volume ratio
- ->Enzymatic system to restore redox environment of cell
- ->Energy production for ion pumps
What are the general clinical finding in hemolytic anemias?
How is the reticulocyte count?
For the diagram: What is “Haem” –>an iron-containing compound of the porphyrin class that forms the nonprotein part of hemoglobin and some other biological molecules.
Clinical Findings
•Jaundice (due to excessive macrophage degradation of Hb that produces bilirubin)
•Scleral icterus (yellowing of the white eye part due to excess bilirubin)
+/- dark urine
+/- splenomegaly (due to excessive breakdown of RBCs)
Laboratory Findings
•↑ Reticulocyte count, RPI
•↑ Lactate dehydrogenase
•↑ Unconjugated bilirubin
•↓ Haptoglobin
•↑ Urobilinogen (bilirubin in urine)
•+/- Hemoglobinuria (Hb in urine), hemoglobinemia (excess Hb in blood plasma)

Difference between extravascular and intravascular hemolysis:
1- Splenomegaly is present in __________, and absent in __________.
2- Dark urine is observed in __________, and not seen on __________.
3- Haptoglobin in low in both hemolysis but its more absent in _______.
4- Hemoglobinuria is present in _________ and absent in________.
5- Spherocytes are seen in _________, and schistocyes and RBC fragments in _________.
1- extravascular; intravascular
2- intravascular; extravascular
3- Intravascular
4- intravascular; extravascular
5- Extravascular; intravascular

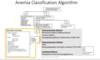
Anemia Classification diagram:
- Describe the major constituents of the RBC membrane and cytoskeleton, identify the major defects in hereditary spherocytosis, and relate these to the clinical and laboratory findings of the disorder.
Hereditary spherocytosis (HS) is a familial disorder characterized by:
- anemia, intermittent jaundice, splenomegaly and responsiveness to splenectomy.
- The heterogeneity of clinical features is associated with multiple molecular abnormalities of which spectrin deficiency is the most common.
- The hallmark of this syndrome is loss of plasma membrane and formation of the microspherocyte. Spherocytes are more susceptible in vitro to osmotic stress, the basis for a common test for the disorder.
- The basic pathophysiology is that spectrin, ankyrin or band 3 defects weaken the cytoskeleton and destabilize the lipid bilayer. Loss of membrane and formation of the spherocyte leads to decreased deformability and entrapment in the spleen. Conditioning in the red pulp leads to further loss of red cell membrane and, ultimately, removal by the macrophage.
Clinically, patients present with a variable degree of anemia as well as jaundice and splenomegaly.
- One third have hyperbilirubinemia as neonates. Most inherit the condition as an **autosomal dominant; 25% as autosomal recessive.
- Treatment includes supportive care for chronic anemia and intermittent complications and splenectomy which usually resolves the clinical manifestations.
What does laboratory results show?
Laboratory features include variable Hct and Hgb (a small group may have no or mild anemia),
-increased reticulocyte count and index
-decreased MCV
- spherocytes on smear
- unconjugated hyperbilirubinemia and an abnormal osmotic fragility test.

Hereditary Spherocytosis
What is the inheritance patter?
What is the presentation of the disease?
What are some common complications?
What is the treatment?

Genetic inheritance pattern: Autosomal dominant >> autosomal recessive
Incidence 1:5000
Extravascular hemolytic anemia (severe 5%, moderate 60-75%, mild 20%)
•Presentation:
–>Neonatal jaundice (because of increased UCB from splenic macrophage destruction of RBCs)
Hemolytic exacerbation
Aplastic crisis
Splenomegaly (due to hypertrophy from RBC hemolysis)
Complications
•Aplastic crisis
•Bilirubin gallstones (Due to increased liver conversion of UCB to conjugated bilirubin, which is excreted in the bile. Conjugated bilirubin is converted back to UCB in the gallbladder and combines with calcium to form the stones).
Treatment
•Folate supplementation
•Splenectomy (anemia resolves)

Diagnosis of spherocytosis:

•Variable anemia
↑ reticulocyte count and RPI
↑ MCHC (only anemia with an increased MCHC, happens due to cellular dehydration from the loss of water and sodium).
- Spherocytes
- ↑ unconjugated bilirubin
- ↑ osmotic fragility
Pyruvate Kinase Deficiency (PKD)
Type of inheritance?
Type of hemolysis?
Treatment?
- Pyruvate kinase deficiency = most common glycolytic pathway defect
- Autosomal recessive inheritance
- Variable primarily extravascular hemolytic anemia
- Splenomegaly
- Bilirubin gallstones
- Treatment
- Splenectomy, transfusions as needed
Glucose-6-Phosphatase Dehydrogenase Deficiency
Type of inheritance?
Where is it prevalent?
What kind of hemolysis?
Clinical presentation?
What is the morphology?

- Inheritance = X-linked
- Prevalent in the Mediterranean, Africa, Asia (endemic malaria areas)
Presentation
•Intermittent episodes of acute hemolytic anemia, ↑ reticulocyte count, unconjugated bilirubin
•Neonatal hyperbilirubinemia
•Primarily *extravascular hemolysis, some intravascular hemolysis
Morphology: Microspherocytes, blister cells, bite cells, Heinz bodies
Triggers: **fava beans, oxidant drugs/foods, infections
Treatment
•Avoidance of oxidant triggers
•Folate supplementation
•Supportive care

Food & Drugs to Avoid in G6PD
Antibiotics containing what should be avoided?
What kind of blue shoulde be avoided?
Mothballs?
- Fava or broad beans
- Aspirin***
- Antimalarials: chloroquine, primiquine, quinidine, quinine
- *Sulfa containing antibiotics
- Nitrofuran antibiotics
- Naphthalene (mothballs)
- Methylene blue
Autoimmune Hemolytic Anemia:
Cold
Cold (referring to a 4°C temperature for maximal in vitro effect) antibodies of IgG or IGM class transiently bind red cell membrane in cooler areas of the body (fingers, toes, ears, skin).
-As they move back to central circulation, they avidly activate complement through the C5-9 attack complex which creates holes in the plasma membrane. When cells move more centrally, the antibody dissociates itself because of low affinity at higher temperatures and complement is left to destroy the cell (intravascular hemolysis).
Autoimmune Hemolytic Anemia
Warm
Who is the main killer in this autoimmune hemolytic anemia?
Warm (maximal effect at 37°C) antibodies, usually IgG, bind the red cell with high affinity and have no or poor complement activating capacity inciting the splenic macrophage to antibody-mediated phagocytosis through the Fc receptor.
Occasionally, the small amount of C3 also induces phagocytosis through complement receptors. Clearance by phagocytosis results in extravascular hemolysis.
Comparisson between warm and cold autoimmune hemolytic disease:
1- Autoantibody Type in Warm is ________, in cold is _________, in paroxysmal cold hemoblobinuria is _________.
2-Antigen specificity in Warm is ________, in cold is _________, in paroxysmal cold hemoblobinuria is _________.
3- Hemolysis in Warm is ________, in cold is _________, in paroxysmal cold hemoblobinuria is _________.
1- IgG, IgM, IgG
2- Panreactive or Rh complex, I antigen, P antigen
3- Extravascular, intravascular, intravascular

Direct Antibody test (Direct Coombs)
Used for detect?
Coombs reagents has antobodies for?
What is the indirect Antibody test>

Antiglobulin or Coombs tests are used to detect IgG and/or complement on the surface of the cell.
The direct antiglobulin test (DAT) evaluates the presence of either IgG or C3d or C4d on the surface of the patient’s red cells by the addition of Coombs reagent which has antibodies for IgG, C3d and C4d causing agglutination.
The indirect antiglobulin detects the ability of patient’s serum to bind IgG and/or complement to test (normal) red blood cells. By definition, autoimmune hemolytic anemia should have a positive DAT

Autoiimune hemolytic Anemia
Treatment for warm AIHA?
Treatment of cold AIHA?
Warm AIHA
•Steroids, treatment of underlying disorder, rituximab (anti-CD20 therapy), splenectomy.
Cold AIHA
•Avoidance of cold
